As more and more people have done advanced Y chromosome tests, we are learning more about how exactly we are related to one another along the male line. Some theories are confirmed, other long held assumptions are turned on their heads.
It is truly an exciting time to be engaged in genetic genealogy given the accessibility of consumer genetic tests and the ever growing body of ancient DNA samples. Now that we all have, at our fingertips, an increasingly large and complex set of data, we can now more than ever benefit from free, publicly available analytical tools to turn data into useful information.
PhyloGeographer
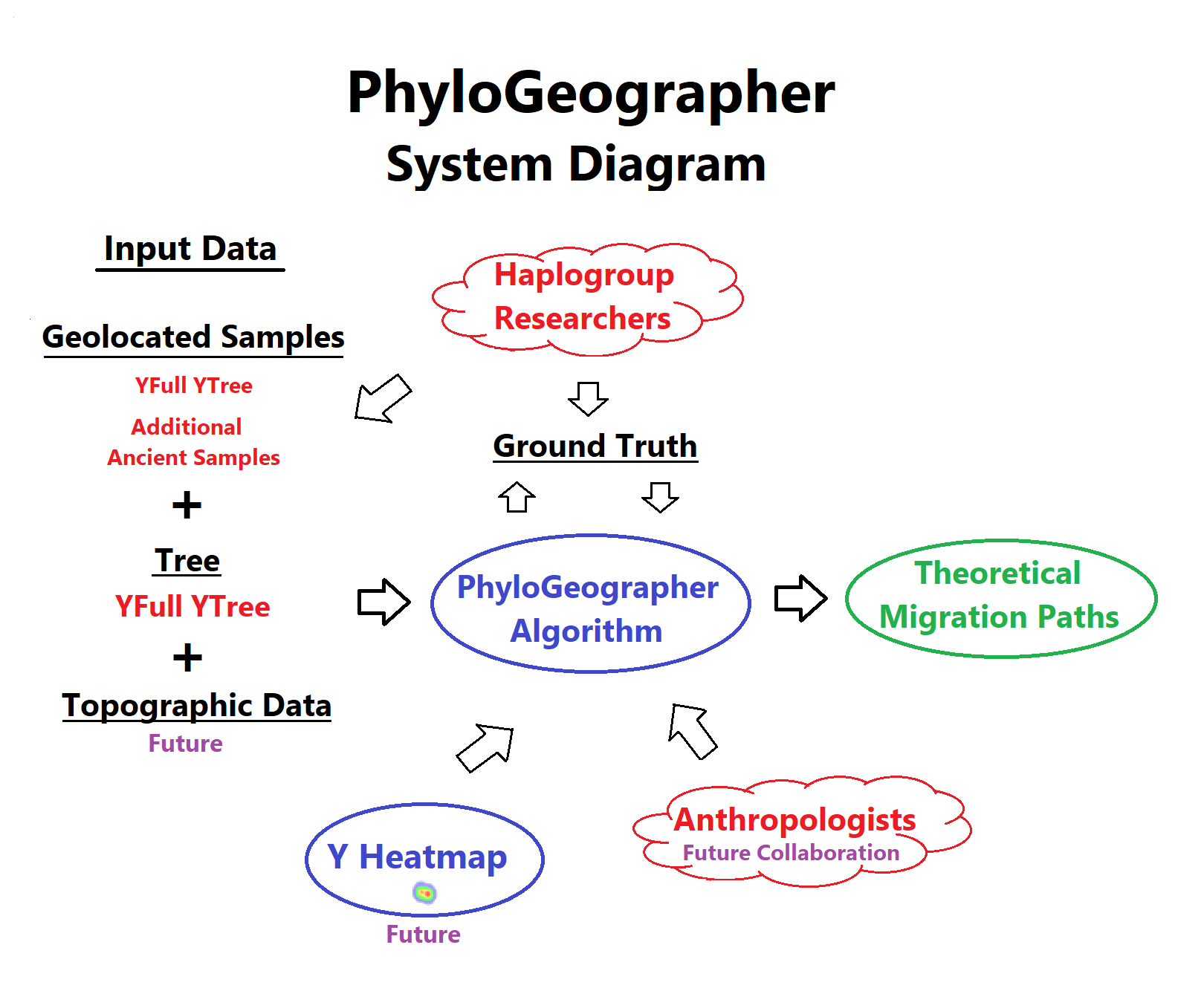
My first contribution to genetic genealogy was PhyloGeographer. Phylogeographer uses an algorithm that takes as input data the geolocated samples on the YFull YTree, additional ancient samples, and the tree structure of the YFull YTree itself along with TMRCA dates. From this input data, the PhyloGeographer algorithm computes a theoretical migration path for every modern haplogroup, starting with Adam.
I created the website in the fall of 2017. Initially each haplogroup's tree structure needed to be hand-written into a CSV file in the appropriate format. This was a lot of work that needed to then be updated each month for each haplogroup. Later, after getting permission from YFull to use their tree, I began to automatically import the YFull YTree. I then improved the algorithm to take the TMRCA date estimates into account (video below).
It's important to understand the realistic goal of the project. The expectation is not to have a crystal ball that automatically displays a migration path that conforms to someone's theory of the "correct" migration for every haplogroup group.
Instead a major goal is the continuous improvement of the algorithm itself so that it does an overall better job of automatically computing theoretical migrations. This project is always a work in progress and instances where a theoretically computed path deviates from a commonly accepted origin, are, depending on the input data, either an opportunity to improve the algorithm by adding to the ground truth set or a window into a possible alternative migration theory worth considering.
There are number difficult problems involved in designing a phylogeography algorithm. Some samples may have reported a completely different paternal ancestor location than their true ancestry. How to consistently determine whether a sample or subclade's computed position should be considered an outlier with respect to its siblings for determination of parent clade origin?
One way to determine which parameters should be used by the algorithm to make this and other decisions, is to define a set of ground truth, against which the algorithm will determine which parameters minimize the amount by which computed paths deviate from it.
I've set up a collaboration system to allow haplogroup researchers to add ancient samples for their haplogroup and have created a spreadsheet for ground truth. I'm hoping that once I've implemented some major upgrades, there will be renewed interest from the haplogroup research community to collaborate to improve the PhyloGeographer system.
Planned upgrades to PhyloGeographer:
- Topographic data integration
- Y Heatmap integration
- Collaboration with anthropologist(s) to improve the algorithm
Y Heatmap
Y Heatmap is a system that computes relative frequency heatmaps for male haplogroups defined by the geolocated samples from the YFull YTree.
It is the result of a collaboration between myself, Thomas Krahn (YSEQ) and YFull.
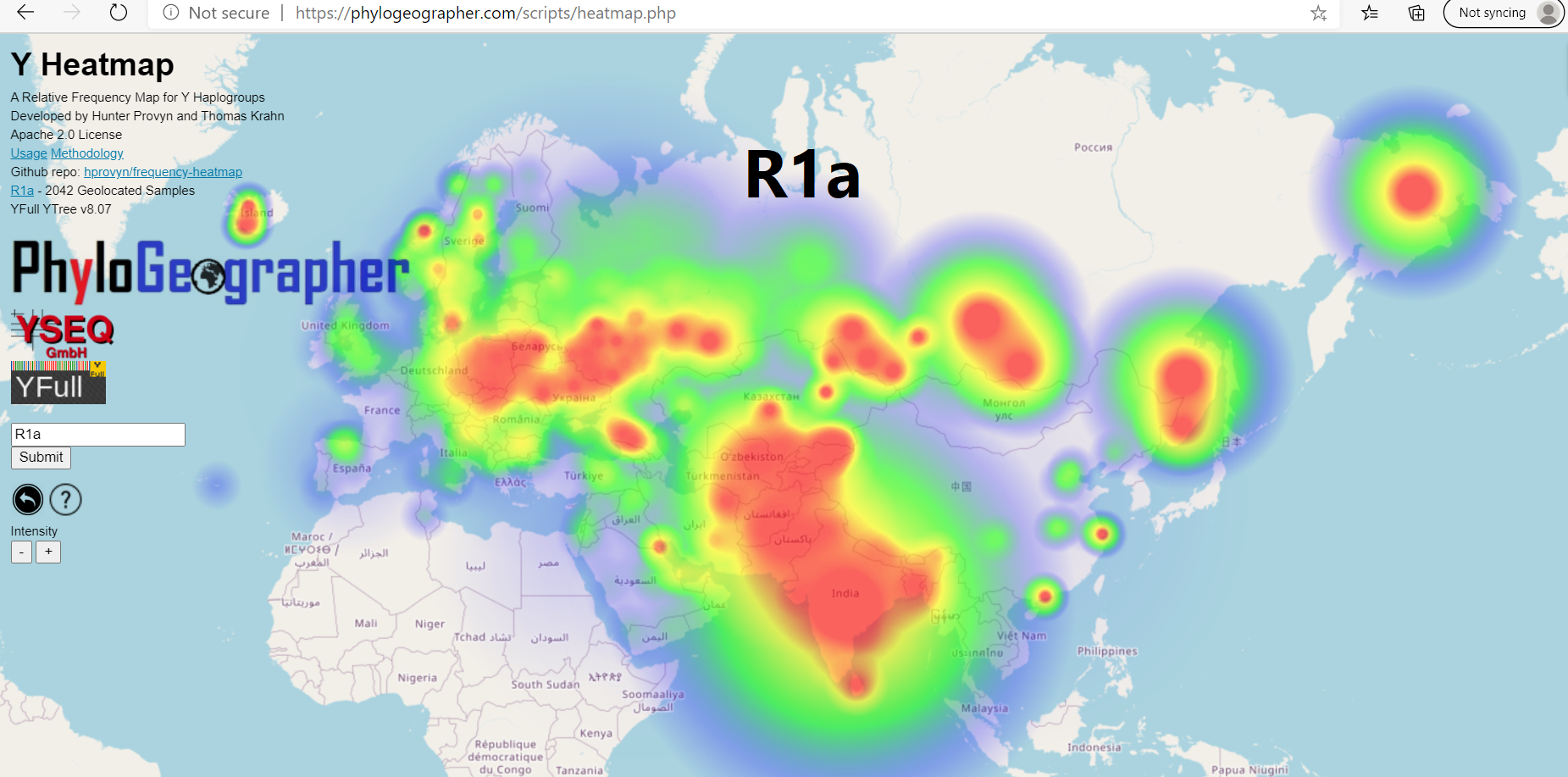
When you zoom closer you can click on the individual samples and see their Clade Finder output. This is a simple visualization of their position on the YFull tree along with links to relevant SNP tests available to order at YSEQ.
In the future I plan to develop a different version of the heatmap that will be immune to founder effects. This type of map would no longer be a true relative frequency heatmap, because on this map the geolocated positions of the offspring of two siblings from 500 years would be given equal weight even if one sibling has ten times as many modern descendants on the YFull tree. I plan to integrate this type of founder effect immune heatmap as a new data source for PhyloGeographer.
Clade Finder
Find your terminal subclade, as best as can be determined from your autosomal test or manual SNP entry. Gateway to your haplogroup's research with links to frequency and migration maps. Quick links to order tests for available phyloequivalent SNPs at YSEQ and recommendations for relevant SNP panels.
Clade Finder is a free tool anyone can use to determine their most specific position on the YFull YTree based on a set of positive and negative SNP calls that can either be entered manually or loaded from one of several file formats used by commercial DNA test vendors (23andMe, AncestryDNA, MyHeritage, VCF).
Based on the positive and negative calls compared against the YFull YTree, a terminal haplogroup is predicted. Positive SNPs displayed in green, negative in red. SNPs with a dollar sign can be ordered at YSEQ to advance your research.

This isn't a really sophisticated analysis tool but it does provide a simple gateway to introduce autosomal testers to their haplogroup. Along with the terminal subclade are a set of links to their position on the YFull tree, their relative frequency heatmap on Y Heatmap and their computed theoretical migration on PhyloGeographer. PhyloGeographer acts as a further gateway to haplogroup research by displaying links to relevant haplogroup research forums/groups.
STR Match Finder
Find distant, yet reliable relatives on the basis of shared rare STR alleles among samples who have joined public projects. Use it to identify samples that are likely to split bottlenecks in your lineage, targeting them for advanced testing to add substructure to your phylogeny.
The inspiration for this tool began with simple python scripts I wrote to find distant yet reliable STR matches that are not provided to FTDNA STR customers. The FTDNA matching interface arranges matches by genetic distance but there are a few problems that sometimes limit its usefulness for finding your closest relatives.
- Low genetic distance (GD) cutoffs. You could be related to someone within several hundred years and not see them as a match.
- No differentiation between stable and unstable STRs. Stable STR differences count for more actual genetic distance.
- No indication of shared rare STR alleles that may be indicative of relatively recent common descent.
The problem of low GD cutoffs is exacerbated in haplogroups like mine, J2, who are less heavily represented in some of the major FTDNA markets.
I now use STR Match Finder almost every day to find distant, reliable matches between those who have tested at YSEQ and FTDNA and joined public projects. You can also import a STR file from YFull.
Recently I've used the tool to discover that a man descending from Zaporozhian Cossacks is likely to be J-Y167175*, with TMRCA 6000 years ago. If he ends up being basal then we see the power of the visual indicators of shared rare alleles for splitting subclades.
No STR data is stored on STR Match Finder - users must enter their project's STRs in a tabular format. I would just create your own private Google Drive doc containing your project's STRs and copy paste it each time you use the tool.
There is an integration with YSEQ to automatically load samples from public groups. For a sample you would like to contact, you may look up the contact info of the group owner.
Thank You
Thank you for being interested in genetic genealogy or haplogroup research and for reading this. If you have done a Next Generation Test like WGS, Big Y, or FGC Y Elite and done the YFull analysis, thank you especially for improving the set of data that underpins my analytical work. You are furthering our knowledge into our common origins.
Thanks to Thomas Krahn of YSEQ for supporting the development of Y Heatmap and Clade Finder. By the way if you are a groups
Thanks to the YFull team for allowing me to use their tree for my analytical tools.
I'm not independently wealthy and devote a significant amount of my time to developing free tools for haplogroup research. If you would like to support my efforts developing free analytical tools consider becoming a patron on Patreon.